
A new international study by scientists at the University of Granada (UGR) in Spain, the University of Wisconsin-Madison, Columbia University in New York, and Rutgers, The State University of New Jersey, has uncovered new data on the coenzyme Q10 (CoQ10), a molecule which is normally synthesized within an organism’s own cells that is essential for cellular metabolism function.
The researchers say this study opens the door for future development of tools designed to modulate CoQ10 synthesis in human cells according to their specific metabolic needs. This ability will be particularly important for treatment of diseases caused by primary and secondary CoQ10 deficiencies.
CoQ10’s role in the energy production required by cells and in their antioxidant capacity, are among the coenzyme’s best known functions. Human cases have been described attributing CoQ10 deficiency to biosynthetic pathway defects, resulting in a syndrome with a distinctly heterogeneous clinical profile.

CoQ10 deficiency is a rare mitochondrial disease affecting mainly children. The composition of the defective biosynthetic pathways is only partially understood because there are elements whose catalyzing enzymes remain unknown, or proteins in the pathway whose specific function are either unknown or have not yet been fully demonstrated.
One such protein is Coq9, which the research group at UGR demonstrated in 2013, and which is an essential protein for CoQ biosynthesis, and for specifically regulating the Coq7 protein, an enzyme with hydroxylase activity that catalyses one of the intermediate steps for CoQ10synthesis.
 The study, currently led by Dr. David J. Pagliarini of the University of Wisconsin-Madison in collaboration with
The study, currently led by Dr. David J. Pagliarini of the University of Wisconsin-Madison in collaboration with  Dr Liang Tong’s team at Columbia U. and UGR researchers Marta Luna Sanchez and Luis Carlos Lopez Garcia was recently published in the journal PNAS.
Dr Liang Tong’s team at Columbia U. and UGR researchers Marta Luna Sanchez and Luis Carlos Lopez Garcia was recently published in the journal PNAS.
 The PNAS paper, entitled “Mitochondrial COQ9 is a lipid-binding protein that associates with COQ7 to enable coenzyme Q biosynthesis“ (PNAS 2014 111 (44) E4697-E4705; published ahead of print October 22, 2014, doi:10.1073/pnas.1413128111), is coauthored by Danielle C. Lohman, Farhad Forouhar, Emily T. Beebe, Matthew S. Stefely, Catherine E. Minogue, Arne Ulbrich, Jonathan A. Stefely, Shravan Sukumar, Marta Luna-Sanchez, Adam Jochem, Scott Lew, Jayaraman Seetharam, Rong Xiao, Huang Wang, Michael S. Westphal, Russell L. Wrobel, John K. Everett, Julie C. Mitchell, Luis C. Lopez, Joshua J. Coon, Liang Tong, and David J. Pagliarini, who say their research “conclusively proves that protein CoQ9 regulates enzyme CoQ7.
The PNAS paper, entitled “Mitochondrial COQ9 is a lipid-binding protein that associates with COQ7 to enable coenzyme Q biosynthesis“ (PNAS 2014 111 (44) E4697-E4705; published ahead of print October 22, 2014, doi:10.1073/pnas.1413128111), is coauthored by Danielle C. Lohman, Farhad Forouhar, Emily T. Beebe, Matthew S. Stefely, Catherine E. Minogue, Arne Ulbrich, Jonathan A. Stefely, Shravan Sukumar, Marta Luna-Sanchez, Adam Jochem, Scott Lew, Jayaraman Seetharam, Rong Xiao, Huang Wang, Michael S. Westphal, Russell L. Wrobel, John K. Everett, Julie C. Mitchell, Luis C. Lopez, Joshua J. Coon, Liang Tong, and David J. Pagliarini, who say their research “conclusively proves that protein CoQ9 regulates enzyme CoQ7.
The researchers note that Coenzyme Q (CoQ), an isoprenylated quinone that is essential for cellular respiration and is synthesized in mitochondria by the combined action of at least nine proteins (COQ19), functions as a requisite component of the mitochondrial oxidative phosphorylation process that is responsible for producing more than 90 percent of cellular Adenosine triphosphate (ATP), a nucleoside triphosphate used in cells as a coenzyme. However, they observe that despite CoQ having been discovered more than 50 years ago, many aspects of its biosynthesis remain obscure, including the functions of uncharacterized CoQ-related proteins whose disruption can cause human disease.
“Our work reveals that one such protein, COQ9, is a lipid-binding protein that enables CoQ biosynthesis through its physical and functional interaction with COQ7, and via its stabilization of the entire CoQ biosynthetic complex,” say the coauthors. “Unexpectedly, COQ9 achieves these functions by repurposing an ancient bacterial fold typically used for transcriptional regulation. Collectively, our work adds new insight into a core component of the CoQ biosynthesis process.”
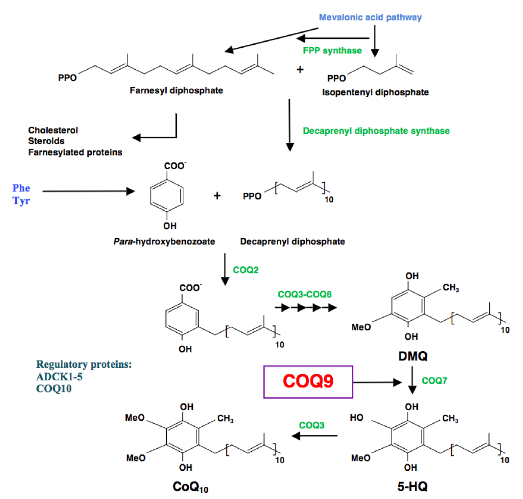
They further observe that although most COQ proteins are known to catalyze CoQ precursor modification, COQ9’s biochemical role remains unclear.
In this paper, the scientists report that a disease-related COQ9 mutation leads to extensive disruption of the CoQ protein biosynthetic complex in a mouse model, and that COQ9 specifically interacts with COQ7 through a series of conserved residue. Toward understanding how COQ9 can perform these functions, they solved the crystal structure of Homo sapiens COQ9 at 2.4, noting that collectively their data define COQ9 as the first, to their knowledge, mammalian TFR structural homolog, suggesting that its lipid-binding capacity and association with COQ7 are key features for enabling CoQ biosynthesis.
The scientists conclude that “crystallization of the human protein and experiments conducted on mice, the study proves that Coq9 has a lipid-binding structure, which would give it the capacity to provide enzyme Coq7 with the intermediary metabolite that it uses as a substrate in the reaction it catalyses.”
The study results suggest additionally that CoQ10’s biosynthetic machinery is organized in mammals as a multi-protein complex with the purpose of increasing its synthesis efficiency and enabling its regulation.
The study’ findings are elaborated on in an accompanying PNAS Plus Significance Statement (vol. 111 no. 44, 15612-15613
DOI10.1073/pnas.ss11144″
Sources:
University of Granada
PNAS
Wikipedia
Image Credits:
University of Granada
University of Wisconsin-Madison
Columbia University